ตำรายาของประเทศไทย
Thai Pharmacopoeia
สำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public Healthสำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public Health1.16 PHARMACEUTICAL DOSAGE FORMS
Dosage forms are provided for most of the Pharmacopoeial drug substances, but the processes for the preparation of many of them are, in general, beyond the scope of the Pharmacopoeia. In addition to defining the dosage forms, this section presents general requirements of some of them. Besides these requirements, the pharmaceutical products should be designed to possess certain desirable properties of bioavailability and stability.
Bioavailability
Bioavailability1 is the rate and extent of absorption of a drug from a dosage form as determined by its concentration/time curve in the systemic circulation or by its excretion in urine. A variety of factors are known to affect absorption. They are, for example, the method of manufacture or method of compounding; the particle size and crystal form or polymorph of the drug substance; and the diluents and excipients used in formulating the dosage form, including fillers, binders, disintegrating agents, lubricants, coatings, solvents, suspending agents, and dyes. Lubricants and coatings are foremost among these. The maintenance of a demonstrably high degree of bioavailability requires particular attention to all aspects of production and quality control that may affect the nature of the finished dosage form.
Stability
Stability of a pharmaceutical dosage form2 refers to the capability of the dosage unit, in a specific container/ closure system, to remain within its physical, chemical, microbiological, therapeutic, and toxicological specifications. The shelf-life of the dosage form is the period of time during which a product is expected, if stored under recommended conditions, to remain within the specification as determined by stability studies on a number of batches of the product. The shelf-life is used to establish the expiration date.
The stability parameters of a pharmaceutical dosage form can be influenced by environmental conditions of storage (temperature, light, air, and humidity), as well as the package components. Pharmacopoeial articles should include required storage conditions on their labelling. These are the conditions under which the expiration date shall apply. The storage requirements specified in the labelling for the article must be observed throughout the distribution of the article (for example, beyond the time it leaves the manufacturer up and including its handling by the dispenser or seller of the article to the consumer).
AEROSOLS
Aerosols are presented in special containers under pressure of a gas and contain one or more active ingredients. The preparations are released from the container, upon actuation of an appropriate valve, in the
1 Further information on bioavailability may be obtained from (1) “ASEAN Guidelines for the Conduct of Bioavailability and Bioequivalence Studies”, issued by ASEAN Pharmaceutical Product Working Group (ASEAN PPWG), 2005; (2) “Instruction for the In Vivo Bioequivalence Study Protocol Development”, notified by Drug Control Division, Food and Drug Administration, Ministry of Public Health, Thailand, 2006.
2 The stability study requirements may be obtained from (1) “ASEAN Guideline on Stability Study on Drug Product”, issued by ASEAN Pharmaceutical Product Working Group (ASEAN PPWG), 2005; (2) “Guidelines for Stability Testing of Pharmaceutical Products”, published by Food and Drug Administration and Department of Medical Sciences, Ministry of Public Health, Thailand, 2004.
form of an aerosol (dispersion of solid or liquid particles in a gas, the size of the particles being adapted to the intended use). The pressure for the release is generated by suitable propellants. The preparations consist of a solution, an emulsion or a suspension and are intended for local application to the skin or the mucous membranes of various body orifices or for inhalation.
The term “aerosol” refers to the fine mist of spray that results from most pressurized systems. However, the term has been broadly misapplied to all self-contained pressurized products, some of which deliver foams or semisolid fluids. The basic components of an aerosol system are the container, the propellant, the concentrate containing the active ingredient(s), the valve, and the actuator. Suitable auxiliary substances may also be used, for example solvents, solubilizers, emulsifying agents, suspending agents and lubricants for the valve to prevent clogging.
Propellants The propellants are either gases liquefied under pressure or compressed gases or low-boiling liquids. Liquefied gases are, for example, halogenated hydrocarbons (especially chloro-fluoro-derivatives of methane and ethane) and low-molecular-mass hydrocarbons (such as propane and butane). Compressed gases are, for example, carbon dioxide, nitrogen and nitrous oxide. Mixtures of these propellants may be used to obtain optimal solution properties and desirable pressure, delivery and spray characteristics.
Valves A suitable valve keeps the container tightly closed when not in use and regulates the delivery of the contents during use. The spray characteristics are influenced by the type of spraying device, in particular by the dimensions, number and location of orifices. Some valves provide a continuous release, others (“metering dose valves”) deliver a defined quantity of product upon each valve actuation. The various valve materials in contact with the contents are compatible with them.
Actuators An actuator is the fitting attached to an aerosol valve stem which, when depressed or moved, opens the valve, and directs the spray containing the drug preparation to the desired area. The actuator usually indicates the direction in which the preparation is dispensed and protects the hand or finger from the refrigerant effects of the propellant. Actuators incorporate an orifice which may vary widely in size and shape. The size of this orifice, the expansion chamber design, and the nature of the propellant and formulation influence the physical characteristics of the spray foam, or stream of solid particles dispensed. For inhalation or oral dose aerosols, an actuator capable of delivering the medication in the proper particle size range is utilized.
Production Aerosols are prepared by one of two general processes. In the “cold-fill” process, the concentrate (generally cooled to a temperature below 0º) and the refrigerated propellant are measured into open containers (usually chilled). The valve-actuator assembly is then crimped onto the container to form a pressure-tight seal. During the interval between propellant addition and crimping, sufficient volatilization of propellant occurs to displace air from the container. In the “pressure-fill” method, the concentrate is placed in the container, and either the propellant is forced under pressure through the valve orifice after the valve is sealed, or the propellant is allowed to flow under the valve cap and then the valve assembly is sealed (“under-the-cap” filling).
In both cases of the “pressure-fill” method, provision must be made for evacuation of air by means of vacuum or displacement with a small amount of propellant. Manufacturing process controls usually include monitoring or proper formulation and propellant fill weight, and pressure testing and leak testing and valve function testing of the finished aerosol. Microbiological attributes should also be controlled.
Leak testing Aerosols comply with the Leak testing under “Test for Aerosols” (Appendix 4.19).
Minimum fill Aerosols comply with the test described in the “Minimum Fill” (Appendix 4.26).
Packaging and storage The containers are tight and resistant to the internal pressure and may be made of metal, glass, plastic or combinations of these materials. They are compatible with their contents. Suitable metals include stainless steel, aluminium, and tin-plated steel. Glass containers are protected with a plastic coating. Aerosols shall be stored at a temperature not exceeding 50º and protected from frost.
Labelling The label of aerosols states (1) the method of use, and, if necessary, that the container should be shaken before use; (2) if necessary, the precautions to be taken, for example, avoid inhaling, avoid contact with the eyes and other mucous membranes; (3) that the container should not be exposed to or stored at a temperature above 50º and should not be exposed to direct sunlight; (4) for a container with a metering dose valve, the amount of active ingredient in a unit spray; (5) that the container should not be punctured or incinerated.
AROMATIC WATERS
Aromatic waters are clear saturated solutions of volatile oils or other aromatic substances in water, usually employed for their flavouring rather than their medicinal properties. Aromatic waters prepared as described below contain a small amount of Ethanol.
Production Aromatic waters are prepared by (1) dilution of a concentrated, ethanolic solution of the aromatic substance with water; (2) solution of the aromatic substance, with or without the use of a dispersing agent; or (3) distillation of the aromatic substance.
Packaging and storage Aromatic waters should be kept in tightly closed containers, protected from intense light and excessive heat.
CAPSULES
Capsules are solid dosage forms with hard or soft shells. They are of various shapes and sizes, and contain a single dose of one or more active ingredients. They are intended for oral administration, but preparations for alternative applications may require a special formulation, method of manufacture, or form of presentation, appropriate to their particular use. For this reason they may not comply with certain sections of this monograph. Starch capsules (often known as cachets) are not described in this monograph.
The different categories of capsules that exist include hard, soft, and modified-release capsules. Their surfaces may bear symbols or other markings. They should be sufficiently robust to withstand handling, including packaging, storage, and transportation, without cracking or breaking. They should be packaged and stored in a manner that protects them from microbial contamination. Capsule shells are made of gelatin or other substances, the consistency of which may be modified by the addition of substances such as glycerol and sorbitol. Preservatives may also be necessary. The shell should disintegrate in the presence of digestive fluids so that the contents are released. The contents of capsules may be solid, liquid or of a paste-like consistency.
Capsule shells and contents may contain excipients such as diluents, sweeteners, colouring matters, flavouring substances, disintegrating agents, glidants, lubricants, and substances capable of modifying the behaviour of the active ingredient(s) in the gastro-intestinal tract. The contents should not cause deterioration of the shell.
When excipients are used, it is necessary to ensure that they do not adversely affect the stability, dissolution rate, bioavailability, safety, or efficacy of the active ingredient(s); there must be no incompatibility between any of the components of the dosage form.
Disintegration Capsules comply with the “Disintegration Test for Tablets and Capsules” (Appendix 4.23). For those capsules for which a dissolution requirement is included in the individual monograph, omission of the requirement for disintegration is considered justifiable and is therefore authorized.
Uniformity of dosage units Unless otherwise prescribed in the individual monographs, capsules comply with the “Uniformity of Dosage Units” (Appendix 4.28).
Visual inspection Unpack and inspect at least 20 capsules. They should be smooth and undamaged. Evidence of physical instability is demonstrated by gross changes in physical appearance, including hardening or softening, cracking, swelling, mottling, or discoloration of the shell.
Packaging and storage Capsules should be kept in well-closed containers at a temperature not exceeding 30º and protected from light, excessive moisture, or dryness.
Hard Capsules
Hard capsules have shells consisting of two prefabricated cylindrical sections that fit together. One end of each section is rounded and closed, and the other is open. The contents of hard capsules are usually in solid form (powder or granules); in certain cases the contents may be in the form of encapsulated powders or micropellets.
Production Hard capsules are prepared by mixing the active ingredient(s) with a number of excipients. Sometimes, the physical characteristics of the mixture allow it to be directly filled into the shell, but it may occasionally be necessary to granulate before filling. Normally the granulate needs to be mixed with lubricants and/or disintegrating agents.
A uniform mass of the capsule mixture is volumetrically fed into the narrower lower section of the shell body which is then closed by slipping the larger section or cap over it. The security of the closure may be ensured by suitable means.
Soft Capsules
Soft capsules have thicker shells than hard capsules, and preservatives are usually added. The shells are of one piece and various shapes. Partial migration of the contents into the shell may occur (and vice versa) depending on the nature of the materials used.
Production Soft capsules are prepared by mixing the active ingredient(s) with a number of excipients. Soft gelatin capsules are usually formed, filled, and sealed in one operation. However, shells for extemporaneous use are sometimes prefabricated. Liquids may be incorporated directly. Solids are usually dissolved or dispersed in a suitable excipient(s) to give a solution or dispersion of thick consistency.
Modified-Release Capsules
Modified-release capsules are hard or soft capsules in which the contents or the shell or both contain additives or are prepared by special procedures such as micro-encapsulation which, separately or together, are designed to modify the rate of release of the active ingredient(s) in the gastro-intestinal tract.
DELAYED-RELEASE CAPSULES (ENTERIC CAPSULES) Delayed-release capsules are hard or soft capsules prepared in such a manner that either the shell or the contents resist the action of the gastric fluid but release the active ingredient(s) in the presence of the intestinal fluid.
All requirements for these specialized dosage forms are given in the individual monographs. EXTENDED-RELEASE CAPSULES Extended-release capsules are designed to slow the rate of release of the active ingredient(s) in the gastro-intestinal tract.
All requirements for these specialized dosage forms are given in the individual monographs. Production Delayed-release capsules are prepared by providing hard or soft capsules with a gastroresistant shell (enteric capsules) or by filling capsules with either granules or particles covered with a gastroresistant coating.
See also under Hard Capsules or Soft Capsules.
EAR PREPARATIONS
Ear preparations are liquid, semi-solid or powder preparations usually containing one or more active ingredients in a suitable vehicle. They are intended for instillation, for spraying, for insufflation, for application to the auditory meatus or as an ear wash. Ear preparations may contain auxiliary substances, for example, to adjust tonicity or viscosity, to adjust or stabilize the pH, to increase the solubility of the active ingredients, to stabilize the preparation or to provide adequate antimicrobial properties. Such additives should not adversely affect the intended medicinal action of the preparation, nor, at the concentrations used, cause toxicity or undue local irritation.
Preparations for application to the injured ear, particularly where the ear-drum is perforated, or prior to surgery are sterile, free from antimicrobial preservatives and supplied in single-unit containers.
Unless otherwise justified and authorized, ear preparations supplied in multiple-unit containers contain a suitable antimicrobial preservative in appropriate concentration, except when the preparation itself has adequate antimicrobial properties. The antimicrobial preservatives should be compatible with the other ingredients of the preparation and should remain effective throughout the period of use of the ear preparations. Five categories of ear preparations may be distinguished: (1) ear drops; (2) ear sprays; (3) semisolid ear preparations; (4) ear powders; (5) ear washes.
Minimum fill Ear preparations comply with the test described in the “Minimum Fill” (Appendix 4.26).
Sterility Where the ear preparations are labelled as sterile, unless otherwise directed in the individual monograph, they comply with the “Sterility Test” (Method I, Appendix 10.1).
Ear Drops
Ear drops are suspensions, emulsions or solutions of one or more active ingredients suspended, dispersed or dissolved in liquids such as water, glycols or fatty oils, suitable for application to the auditory meatus without exerting harmful pressure on the ear-drum. They may also be placed in the auditory meatus by means of a plug impregnated with the liquid. Suspended solids may separate slowly on standing but are easily redispersed on shaking. The size of the dispersed particles should be controlled.
Ear drops are usually supplied in multiple-unit containers fitted with an appropriate applicator.
Containers Ear drops are supplied in containers of glass or suitable plastic that are fitted with an integral dropper or with a screw cap of suitable materials incorporating a dropper and rubber or plastic teat. Alternatively, such a cap assembly is supplied separately.
Labelling The label of ear drops states (1) the name(s) and concentration(s) of the active ingredient(s); (2) the name(s) and concentration(s) of any antimicrobial preservative(s); (3) that they are intended for external use only; (4) where appropriate, that the preparation is sterile.
Ear Sprays
(Note When ear sprays are supplied in aerosol containers, these comply with the appropriate requirements for Aerosols.)
Ear sprays are suspensions, emulsions or solutions of one or more active ingredients suspended, dispersed or dissolved in liquids suitable for spraying to auditory meatus without exerting harmful pressure on the eardrum. The special requirements may be necessary for the selection of propellants, for particle size for the single-dose delivered by the metering valves. See also under Ear Drops.
Packaging and storage Ear sprays are supplied in multiple-unit containers fitted with an appropriate applicator.
Labelling When ear sprays are supplied in aerosol containers, the label shall state (1) the method of use and, if necessary, that the container should be shaken before use; (2) if necessary, the precautions to be taken, for example, avoid inhaling; (3) that the container shall not be exposed to or stored at a temperature above 50º and should not be exposed to direct sunlight; (4) for a container with a metering dose valve, the amount of active ingredient in a unit-spray; (5) that the container should not be punctured or incinerated.
See also under Ear Drops.
Semi-Solid Ear Preparations
(Note Semi-solid ear preparations comply with the appropriate requirements for Topical Semi-solid Preparations.)
Semi-solid ear preparations are semi-solid dosage forms such as creams, gels, or ointments, etc. intended for application to the external auditory meatus, if necessary by means of a plug impregnated with the preparation. Semi-solid ear preparations are supplied in containers fitted with a suitable applicator.
Labelling See under Ear Drops.
Ear Powders
(Note Ear powders comply with the appropriate requirements for powders.)
Ear powders are fine powders intended for application or insufflation to the external auditory meatus.
Containers Ear powders are supplied in containers fitted with a suitable device for application or insufflation.
Labelling See under Ear Drops.
Ear Washes
Ear washes are solutions intended to cleanse the external auditory meatus. They are usually aqueous solutions with a pH within physiological limits.
See also under Ear Drops.
Containers Ear washes are supplied in containers fitted with a suitable applicator.
Labelling See under Ear Drops.
EXTRACTS
Extracts are preparations of liquid, solid or semisolid consistency, obtained from herbal or animal matter, which is usually dried. Extracts may be subjected to purification processes that increase the content of characterized constituents with respect to the content of dry extractable matter from that which would be expected from extraction with the stated solvent: such extracts are termed “enriched”.
Three types of extract can be distinguished:
Type A Type A extracts (standardized extracts) are adjusted to a defined range of therapeutically active constituents. Standardization is achieved by adjustment of the extract with inert material or by blending extracts.
Type B Type B extracts (quantified extracts) are adjusted to a defined range of active constituents. Adjustments are made either by blending batches of extracts or by blending batches of herbal or animal matter prior to extraction.
Type C Type C extracts are essentially defined by the production process (state of the matter to be extracted, solvent, extraction conditions). Constituents considered to be relevant markers may be determined.
Production Extracts are prepared by maceration, percolation or other suitable validated methods using ethanol or other suitable solvent. The matter to be extracted may undergo a preliminary treatment, for example, inactivation of enzymes, grinding or defatting. In addition, unwanted matter may be removed, if necessary, after extraction. Herbal drugs, animal matters and organic solvents used for the preparation of extracts comply with any relevant monograph of the Pharmacopoeia. For soft and dry extracts where the organic solvent is removed by evaporation, recovered or recycled solvent may be used, provided that the recovery procedures are controlled and monitored to ensure that solvents meet appropriate standards before reuse or admixture with other approved materials.
Water used for the preparation of extracts is of suitable quality. Except for the test for bacterial endotoxins, water complying with the section on Purified Water in bulk of the monograph on Purified Water is suitable. Potable water may be suitable if it complies with a defined specification that allows the consistent production of a suitable extract.
Where applicable, concentration to the intended consistency is carried out using suitable methods,usually under reduced pressure, and at a temperature at which deterioration of the constituents is reduced to a minimum. Volatile oils that have been distilled during processing may be restored to the extracts at an appropriate stage in the manufacturing process. Suitable inert excipients may be added at the various stages of the manufacturing process to improve technological qualities like homogeneity, consistency or stability of active constituents. Where applicable, as a result of analysis of the herbal or animal matter used for the production of extracts, tests for microbiological quality, heavy metals, aflatoxins, and pesticide residues in the extracts have to be carried out.
Labelling For Type A, the label on the container states (1) the herbal or animal matter used; (2) whether the extract is dry, soft or liquid; (3) the composition of the extraction solvent; (4) where applicable, that fresh herbal or animal matter has been used; (5) where applicable, that the extract is “enriched”; (6) the name and amount of any excipient used. For Types B and C, the label on the container states (1) to (6) as for type A; (7) the content of constituents (markers) used for quantification and (8) the range of starting material: final extract (Drug:Extract Ratio or DER).
Liquid Extracts
Liquid extracts are liquid preparations of which, in general, one part by mass or volume is equivalent to one part by mass of the original dried herbal or animal matter. These preparations are adjusted, if necessary, so that they satisfy the requirements for content of solvent, and, where applicable, for constituents or dry residue.
Production Liquid extracts are prepared by using ethanol of suitable concentration or water to extract the stated herbal or animal matter or by dissolving a soft or dry extract (which has been produced using the same strength of extraction solvent as is used in preparing the liquid extract by direct extraction) of the stated herbal or animal matter in either ethanol of suitable concentration or water and filtering, if necessary. A slight sediment may form on standing, which is acceptable as long as the composition of the liquid extract is not changed significantly. Liquid extracts may contain suitable antimicrobial preservatives.
Relative density Where applicable, the liquid extract complies with the limits prescribed in the monograph.
Ethanol content For ethanolic liquid extracts, carry out the “Determination of Ethanol” (Appendix 6.5). The preparation complies with the limits prescribed in the monograph.
Methanol and 2-propanol Not more than 0.05 per cent v/v of methanol and not more than 0.05 per cent v/v of 2-propanol for ethanolic liquid extracts unless otherwise prescribed.
Dry residue In a flat-bottomed dish about 50 mm in diameter and about 30 mm in height, introduce rapidly 2.0 g or 2.0 ml of the extract to be examined. Evaporate to dryness on a water-bath and dry at 105º for 3 hours. Allow to cool in a desiccator over phosphorus pentoxide desiccant or self-indicating silica gel and weigh. Calculate the result as a percentage or in grams per litre.
Packaging and storage Liquid extracts should be kept in well-closed containers, protected from light.
Labelling The label on the container states in addition to the requirements listed above (1) where applicable, the ethanol content in per cent v/v in the final extract; (2) the concentration of any added antimicrobial preservative.
Soft Extracts
Soft extracts are semi-solid preparations obtained by evaporation of the solvent used for preparation. Soft extracts generally have a dry residue of not less than 70 per cent w/w. They may contain suitable antimicrobial preservatives.
Dry residue In a flat-bottomed dish about 50 mm in diameter and about 30 mm in height, weigh rapidly 2.0 g of the extract to be examined. Heat to dryness on a water-bath and dry at 105º for 3 hours. Allow to cool in a desiccator over phosphorus pentoxide desiccant or selfindicating silica gel and weigh. Calculate the result as a percentage weight in weight. Where applicable, a monograph on a soft extract prescribes a limit test for the solvent used for extraction.
Packaging and storage Soft extracts should be kept in well-closed containers, protected from light.
Labelling The label on the container states in addition to the requirements listed above (1) where applicable, the ethanol content in per cent v/v in the final extract; (2) the concentration of any added antimicrobial preservative.
Dry Extracts
Dry extracts are solid preparations obtained by evaporation of the solvent used for their production. Dry extracts generally have a dry residue of not less than 95 per cent w/w.
Loss on drying Where applicable, the dry extract complies with the limits prescribed in the monograph. In a flat-bottomed dish about 50 mm in diameter and about 30 mm in height, weigh rapidly 500 mg of the extract to be examined, finely powdered. Dry at 105º for 3 hours. Allow to cool in a desiccator over phosphorus pentoxide desiccant or self-indicating silica gel and weigh. Calculate the result as a percentage weight in weight. Where applicable, a monograph on a drug extract prescribes a limit test for the solvent used for extraction.
Packaging and storage Dry extracts should be kept in well-closed containers, protected from light.
EYE PREPARATIONS
Eye preparations are sterile liquid, semi-solid or solid preparations intended for administration upon the eyeball and/or to the conjunctiva, or for insertion in the conjunctival sac. Several categories of eye preparations may be distinguished: (1) eye drops; (2) eye lotions; (3) powders for eye drops and powders for eye lotions; (4) semi-solid eye preparations; (5) ocular systems.
Production During the development of an eye preparation, whose formulation contains an antimicrobial preservative, the necessity for and the efficacy of the chosen preservative shall be demonstrated to the satisfaction of the competent authority. A suitable test method together with criteria for judging the preservative properties of the formulation are provided in “Efficacy of Antimicrobial Preservation” (Appendix 10.6).
Eye preparations are prepared using materials and methods designed to ensure sterility and to avoid the introduction of contaminants and the growth of microorganisms; recommendations on this aspect are provided in “Sterilization and terility Assurance” (Appendix 12).
In the manufacture of eye preparations containing dispersed particles, measures are taken to ensure a suitable and controlled particle size with regard to the intended use.
For multiple-unit containers of eye preparations, the period after opening the container, after which the contents must not be used, does not exceed 4 weeks.
Sterility Eye preparations comply with the “Sterility Test” (Appendix 10.1). Applicators supplied separately also comply with the test for sterility. Remove the applicator with aseptic precautions from its package and transfer it to a tube of culture medium so that it is completely immersed. Incubate and interpret the results as described in the test for sterility.
Minimum fill Eye preparations except eye strips and ocular systems comply with the test described in the “Minimum Fill” (Appendix 4.26).
Packaging and storage Eye preparations should be kept in a sterile, tightly closed, tamper-evident container.
Labelling The label of eye preparations states the name(s) of any added antimicrobial preservative(s).
Eye Drops
Eye drops are sterile aqueous or oily solutions, emulsions or suspensions of one or more active substances intended for instillation into the eye. Eye drops may contain excipients, for example, to adjust the tonicity or the viscosity of the preparation, to adjust or stabilize the pH, to increase the solubility of the active substance, or to stabilize the preparation. These substances do not adversely affect the intended medicinal action or, at the concentrations used, cause undue local irritation.
Aqueous preparations supplied in multidose containers contain a suitable antimicrobial preservative in appropriate concentration except when the preparation itself has adequate antimicrobial properties. The antimicrobial preservative chosen must be compatible with the other ingredients of the preparation and must remain effective throughout the period of time during which eye drops are in use.
If eye drops are prescribed without antimicrobial preservatives, they are supplied wherever possible in single-dose containers. Eye drops intended for use in surgical procedures do not contain antimicrobial preservatives and are supplied in single-dose containers.
Eye drops that are solutions, examined under suitable conditions of visibility, are practically clear and practically free from particles.
Eye drops that are suspensions may show a sediment that is readily redispersed on shaking to give a suspension which remains sufficiently stable to enable the correct dose to be delivered.
Multidose preparations are supplied in containers that allow successive drops of the preparation to be administered. The containers contain at most 10 ml of the preparation.
Limit of particle size Unless otherwise specified in the individual monograph, eye drops in the form of a suspension comply with the following test. Introduce a suitable quantity of the suspension into a counting cell or with a micropipette onto a slide, as appropriate, and scan under a microscope an area corresponding to 10 μg of the solid phase. For practical reasons, it is recommended that the whole sample be first scanned at low magnification (e.g., ×50) and particles greater than 25 μm are identified. These larger particles can then be measured at a larger magnification (e.g., ×200 to ×500). For each 10 μg of solid active substance, not more than 20 particles have a maximum dimension greater than 25 μm, and not more than 2 of these particles have a maximum dimension greater than 50 μm. None of the particles has a maximum dimension greater than 90 μm.
Labelling The label of eye drops states the storage condition.
Eye Lotions
Eye lotions are sterile aqueous solutions intended for use in rinsing or bathing the eye or for impregnating eye dressings.
Eye lotions may contain excipients, for example to adjust the tonicity or the viscosity of the preparation or to adjust or stabilize the pH. These substances do not adversely affect the intended action or, at the concentrations used, cause undue local irritation.
Eye lotions supplied in multidose containers contain a suitable antimicrobial preservative in appropriate concentration except when the preparation itself has adequate antimicrobial properties. The antimicrobial preservative chosen is compatible with the other ingre-dients of the preparation and remains effective throughout the period of time during which the eye lotions are in use.
If eye lotions are prescribed without an antimicrobial preservative, they are supplied in single-dose containers. Eye Lotions intended for use in surgical procedures or in first-aid treatment do not contain an antimicrobial preservative and are supplied in singledose containers.
Eye lotions, examined under suitable conditions of visibility, are practically clear and practically free from particles.
The containers for multidose preparations do not contain more than 200 ml of eye lotion, unless otherwise specified in the individual monograph.
Labelling See under Eye Drops.
Powders for Eye Drops and Eye Lotions
Powders for the preparation of eye drops and eye lotions are supplied in a dry, sterile form to be dissolved or suspended in an appropriate liquid vehicle at the time of administration. They may contain excipients to facilitate dissolution or dispersion, to prevent caking, to adjust the tonicity, to adjust or stabilize the pH or to stabilize the preparation.
After dissolution or suspension in the prescribed liquid, they comply with the requirements for eye drops or eye lotions, as appropriate.
Uniformity of dosage units Single-unit powders for eye-drops and eye lotions comply with the “Uniformity of Dosage Units” (Appendix 4.28).
Semi-solid Eye Preparations Semi-solid eye preparations are sterile ointments, creams or gels intended for application to the conjunctiva. They contain one or more active substances dissolved or dispersed in a suitable basis. They have a homogeneous appearance. Semi-solid eye preparations comply with the requirements of the monograph on Topical Semi-solid Preparations. The basis is non-irritant to the conjunctiva.
Semi-solid eye preparations are packed in small, sterilized collapsible tubes fitted or provided with a sterilized cannula and having a content of not more than 10 g of the preparation. The tubes must be wellclosed to prevent microbial contamination.
Semi-solid eye preparations may also be packed in suitably designed single-dose containers. The containers, or the nozzles of tubes, are of such a shape as to facilitate administration without contamination.
Limit of particle size Semi-solid eye preparations containing dispersed solid particles comply with the following test: spread gently a quantity of the preparation corresponding to at least 10 μg of solid active substance as a thin layer. Scan under a microscope the whole area of the sample. For practical reasons, it is recommended that the whole sample is first scanned at a small magnification (e.g., ×50) and particles greater than 25 μm are identified. These larger particles can then be measured at a larger magnification (e.g., ×200 to ×500). For each 10 μg of solid active substance, not more than 20 particles have a maximum dimension greater than 25 μm, and not more than 2 of these particles have a maximum dimension greater than 50 μm. None of the particles has a maximum dimension greater than 90 μm.
Labelling See under Eye Drops.
Ocular Systems
See under Systems.
GRANULES
Granules are preparations consisting of solid, dry aggregates of powder particles sufficiently resistant to withstand handling. They are intended for oral administration. Some are swallowed as such, some are chewed and some are dissolved or dispersed in water or another suitable liquid before being administered. Granules contain one or more active ingredients with or without added substances including, where necessary, authorized colouring matter and flavouring agents. Granules are presented as single-unit or multiple-unit preparations. For single-unit preparations each dose is enclosed in an individual container, for example, a sachet, a paper packet or a vial. Each dose of a multiple-unit preparation is administered by means of a device suitable for measuring the quantity prescribed.
Several categories of granules may be distinguished: (1) uncoated granules; (2) granules for the preparation for oral liquids (see under Oral Liquids); (3) coated granules; (4) modified-release granules.
Uniformity of dosage units Unless otherwise prescribed in the individual monographs, granules comply with the “Uniformity of Dosage Units” (Appendix 4.28). The test for Content Uniformity is not required for multivitamin and trace element granules.
Packaging and storage Unless otherwise specified the individual monograph. Granules shall be kept in tightly closed containers.
Labelling For single-unit containers the label states the name(s) and amount(s) of active ingredient(s) per container and for multiple-unit containers the label states the name(s) and amount(s) of active ingredient(s) in a suitable quantity by weight.
Uncoated Granules
Uncoated granules may be plain or effervescent granules.
EFFERVESCENT GRANULES Effervescent granules are uncoated granules generally containing acid substances and either carbonates or bicarbonates which react rapidly in the presence of water to release carbon dioxide. They are intended to be dissolved or dispersed in water before administration.
Disintegration Place a single dose of the granules in a beaker containing 200 ml of water at 15º to 25º; numerous gas bubbles are evolved. When the evolution of gas around the individual grains has ceased, the granules have disintegrated, being either dissolved or dispersed in the water. Repeat the operation on a further five doses. The granules comply with the test if each of the six doses used in the test disintegrates within 5 minutes.
Coated Granules
Coated granules are granules covered with one or more layers of mixtures of various substances. Substances used as coatings are usually applied as a solution or suspension in conditions in which evaporation of the vehicle occurs. Coated granules are usually presented as multiple-unit preparations.
Modified-release Granules
DELAYED-RELEASE GRANULES (ENTERIC-COATED GRANULES) Delayed-release granules are intended to resist the gastric fluid and to release the active ingredient(s) in the intestinal fluid. These properties are achieved by covering the granules with a gastro-resistant material (enteric-coated granules) or by other suitable means.
EXTENDED-RELEASE GRANULES Extended-release granules are coated or uncoated granules prepared by using added substances or procedures which, separately or together, are designed to modify the rate or the place at which the active ingredient(s) are released.
HERBAL DRUG PREPARATIONS
Herbal drug preparations are obtained by subjecting herbal drugs to treatments such as extraction, distillation, expression, fractionation, purification, concentration or fermentation. These include comminuted or powdered herbal drugs, tinctures, extracts, essential oils, expressed juices and processed exudates.
Herbal teas comply with the monograph on Herbal teas.
Instant herbal teas consist of powder or granules of one or more herbal drug preparation(s) intended for the preparation of an oral solution immediately before use.
HERBAL TEAS
Herbal teas consist exclusively of one or more herbal drugs intended for oral aqueous preparations by means of decoction, infusion or maceration. The preparation is prepared immediately before use.
Herbal teas are usually supplied in bulk form or in sachets.
The herbal drugs used comply with the appropriate individual monographs.
Recommendations on the microbiological quality of herbal teas under the “Limits for Microbial Contamination” (Category 2 in Table 2, Appendix 10.5) taking into account the prescribed preparation method (use of boiling or non-boiling water).
Identification The identity of herbal drugs present in herbal teas is checked by botanical examinations.
The proportion of herbal drugs present in herbal teas is checked by appropriate methods. Herbal teas in sachets comply with the following test:
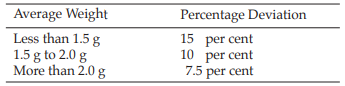
Weight variation Determine the average weight of twenty randomly chosen units as follows: weigh a single full sachet of herbal tea, open it without losing any fragments. Empty it completely using a brush. Weigh the empty sachet and calculate the mass of the contents by subtraction. Repeat the operation on the nineteen remaining sachets. Unless otherwise justified not more than two of the twenty individual masses of the contents deviate from the average mass of the contents by more than the percentage deviation shown in the table below and none deviates by more than twice that percentage.
Packaging and storage Herbal teas should be protected from light.
INFUSIONS
Infusions are dilute solutions that contain the readily soluble constituents of crude drugs. Fresh infusions are made by pouring boiling water onto the drug, in a suitable state of comminution, and macerating for a short time, or they are usually prepared by diluting one volume of a concentrated infusion to ten volumes with water. Concentrated infusions are usually made by maceration of the drug with Ethanol (25 Per Cent).
For dispensing purposes, Infusions should be used within 12 hours of preparation from concentrated infusions.
Packaging and storage Infusions should be kept in well-closed containers.
Labelling The label on the container states the storage conditions.
IRRIGATION SOLUTIONS
Irrigation solutions are sterile solutions of one or more active ingredients intended for irrigation. If the solution is intended to be used for the irrigation of body cavities, for the flushing of wounds or operation cavities or for the irrigation of the urogenital system, it is sterile and apyrogenic. Such solutions are prepared using Water for Irrigation.
Irrigation solutions may contain added substances such as suitable substances to make the preparation isotonic with blood. They are supplied in containers holding sufficient of the solution for use on one occasion only. When viewed under suitable conditions of visibility, they are practically clear and practically free from particles.
Sterility Unless otherwise directed in the individual monograph, irrigation solutions comply with the “Sterility Test” (Method I, Appendix 10.1).
Packaging and storage Irrigation solutions are supplied in containers made from materials that are sufficiently transparent to permit the visual inspection of the contents and that do not cause deterioration of the preparation as a result of diffusion into or across the material of the container or by yielding foreign substances into the preparation. The containers should be readily distinguishable from containers for preparations intended for parenteral administration. The containers are tamper-evident and are sealed so as to exclude micro-organisms. Unless otherwise specified in the individual monograph, Irrigation Solutions should be stored at a temperature not exceeding 25º.
Labelling The label of irrigation solutions states (1) that the irrigation solution is sterile and, where applicable, that the irrigation solution is apyrogenic; (2) that the irrigation solution is not to be used for injection; (3) that the irrigation solution should be used on one occasion only and that any remainder should be discarded.
LOZENGES
See under Oromucosal Preparations.
MEDICATED FOAMS
Medicated foams are preparations consisting of large volumes of gas dispersed in a liquid generally containing one or more active ingredients, a surfactant ensuring their formation and various other excipients. Medicated foams are usually intended for application to the skin or mucous membranes. Medicated foams are usually formed at the time of administration from a liquid preparation in a pressurized container. The container is equipped with a device consisting of a valve and a push button suitable for the delivery of the foam. Medicated foams intended for use on severely injured skin and on large open wounds are sterile. Medicated foams supplied in pressurized containers comply with the requirements in the monograph for “Aerosols” (Appendix 1.16).
Production Sterile medicated foams are prepared using materials and methods designed to ensure sterility and to avoid the introduction of contaminants and the growth of micro-organisms.
Relative foam density Maintain the container at about 25º for at least 24 hours. Taking care not to warm the container, fit a rigid tube 70 mm to 100 mm long and about 1 mm in internal diameter onto the push button. Shake the container to homogenize the liquid phase of the contents and dispense 5 ml to 10 ml of foam to waste. Tare a flat-bottomed dish of about 60 ml volume and about 35 mm high. Place the end of the rigid tube attached to the push button in the corner of the dish, press the push button and fill the dish uniformly, using a circular motion. After the foam has completely expanded, level off by removing the excess foam with a slide. Weigh. Determine the weight of the same volume of water by filling the same dish with water. The relative foam density is equivalent to the ratio:
m/e,
where m is the weight of test sample of foam in g and e is the weight of same volume of water in g. Carry out three measurements. None of the individual values deviate by more than 20 per cent from the mean value.
Duration of expansion The apparatus (Fig. 1) consists of a 50-ml burette, 15 mm in internal diameter, with 0.1-ml graduations and fitted with a 4-mm single bore stopcock. The graduation corresponding to 30 ml is at least 210 mm from the axis of the stopcock. The lower part of the burette is connected by means of a plastic tube not longer than 50 mm and 4 mm in internal diameter to the foam-generating container equipped with a push button fitted to this connection. Maintain the container at about 25º for at least 24 hours. Shake the container, taken care not to warm it, to homogenize the liquid phase of the contents and dispense 5 ml to 10 ml of the foam to waste.
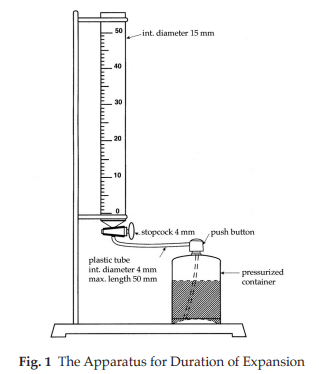
Connect the push button to the outlet of the burette. Press the button and introduce about 30 ml of foam in a single delivery. Close the stopcock and at the same time start the chronometer and read the volume of foam in the burette. Every 10 seconds read the growing volume until the maximum volume is reached. Carry out three measurements. None of the times needed to obtain the maximum volume is more than 5 minutes.
Sterility Comply with the “Sterility Test” (Appendix 10.1), when the label indicates that the preparation is sterile.
Labelling The label on the container states (1) that the Medicated Foam is intended for external use only and (2) the storage conditions.