ตำรายาของประเทศไทย
Thai Pharmacopoeia
สำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public Healthสำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public HealthMethod C
The potency of anti-Rh0 (D) immunoglobulin is determined by flow cytometry in a microtitre plate format. The method is based on the specific binding between anti-Rh0 (D) immunoglobulin and Rh0 (D)- positive red blood cells. The activity of the preparation to be examined is compared with a reference preparation calibrated in International Units.
MATERIALS
Reagents not specified are of analytical grade.
PBS Dissolve 8.0 g of sodium chloride, 0.76 g of disodium hydrogenphosphate, 0.2 g of potassium chloride and 0.2 g of potassium dihydrogenphosphate in water and dilute to 1000 ml with the same solvent.
PBS-BSA solution PBS containing 1 per cent w/v of bovine serum albumin.
Red blood cells Use Rh0 (D)-positive red blood cells obtained from a group OR1R1 donor within 2 weeks of collection. Store if necessary in an appropriate stabilizer at 4º. Wash the cells at least twice with PBS-BSA solution and prepare a suspension containing 1 × 104 cells per microlitre but not more than 5 × 104 cell per microlitre in PBS-BSA solution.
Use Rh0 (D)-negative red blood cells obtained from a group Orr donor and prepared similarly.
Secondary antibody Use a suitable fluorescent dye conjugated anti-IgG antibody-fragment specific for human IgG or parts of it. Store and use according to the manufacturer’s instructions.
Microtitre plates Use flat-bottomed plates without surface treatment for enzyme immunoassays.
METHOD
Test solutions For freeze-dried preparations, reconstitute as stated on the label. Prepare at least three independent replicates of at least three serial 1.5 or twofold dilutions starting with a concentration in the range of 1.2 to 0.15 IU per ml using PBS-BSA solution as diluent. If necessary, adjust the starting dilution to obtain responses falling in the liner portion of the doseresponse curve.
Reference solutions Reconstitute the reference preparation according to instructions. Prepare at least three independent replicates of at least three serial 1.5 or twofold dilutions starting with a concentration in the range of 1.2 to 0.15 IU per ml using PBS-BSA solution as diluent. If necessary, adjust the starting dilutions to obtain responses falling in the linear portion of the dose-response curve.
Distribute 50 μl of the Rh0 (D)-positive red blood cells into each well of a microtitre plate. Add 50 μl of each of the dilutions of the test solutions or reference solution to each of a series of wells. Use 50 μl of PBSBSA solution as negative control. Distribute 50 μl of the Rh0 (D)-negative red blood cells into four wells of the same microtitre plate and add 50 μl of the lowest dilution of the test preparation. To monitor spurious reactions distribute 50 μl of the Rh0 (D)-positive red blood cells into four wells of the same microtitre plate and add 50 μl of PBS-BSA solution. Seal with plastic film and incubate at 37º for 40 minutes.
Centrifuge the plates at 50 × g for 3 minutes, discard the supernatant and wash the cells with 200 to 250 μl of PBS-BSA solution. Repeat this at least once.
Centrifuge the plates at 50 × g for 3 minutes, discard the supernatant and add 50 μl of the secondary antibody diluted with PBS-BSA solution to a suitable protein concentration. Seal with plastic film and incubate, protected from light, at room temperature for 20 minutes.
Centrifuge the plates at 50 × g for 3 minutes, discard the supernatant and wash the cells with 200 to 250 μl of PBS-BSA solution. Repeat this at least once.
Centrifuge the plates at 50 × g for 3 minutes, resuspend the cells into 200 to 250 μl of PBS. Transfer the cell suspension into a tube suitable for the flow cytometry equipment available and further dilute by adding PBS to allow a suitable flow rate.
Proceed immediately with measurement of the median fluorescence intensity in a flow cytometer. Record at least 10,000 events without gating but excluding debris.
Use the median fluorescence intensity in the linear range of the dose response curve to estimate the potency of the preparation to be examined by the “Statistical Analysis of Results of Biological Assays and Test” (Appendix 9).
15.1.7 Biological Assay of Human Coagulation Factor II
Human coagulation factor II is assayed following specific activation to form factor IIa. Factor IIa is estimated by comparing its activity in cleaving a specific chromogenic peptide substrate with the same activity of the International Standard or of a reference preparation calibrated in International Units.
The chromogenic assay method consists of two steps: snake venom-dependent activation of factor II, followed by enzymatic cleavage of a chromogenic factor IIa substrate to form a chromophore that can be quantified spectrophotometrically. Under appropriate assay conditions, there is a linear relation between factor IIa activity and the cleavage of the chromogenic substrate.
REAGENTS
Viper venom specific factor II activator (ECARIN) A protein derived from the venom of the saw-scaled viper (Echis carinatus) which specifically activates factor II. Reconstitute according to the manufacturer’s instructions. Store the reconstituted preparation at 4º and use within 1 month.
Factor II a chromogenic substrate Specific chromogenic substrate for factor IIa such as: H-D phenylalanyl-L-pipecolyl-L-arginine-4-nitroanilide dihydrochloride, 4-toluenesulfonyl-glycyl-prolyl-Larginine-4-nitroanilide, H-D-cyclohexylglycyl-αaminobutyryl-L-arginine-4-nitroanilide, Dcyclohexylglycyl-L-alanyl-L-arginine-4-nitroanilide diacetate. Reconstitute according to the manufacturer’s instructions.
Dilution buffer Solution containing 0.606 per cent w/v of tris(hydroxymethyl)aminomethane, 1.753 per cent w/v of sodium chloride, 0.279 per cent w/v of (ethylenedinitrilo)tetra-acetic acid and 0.1 per cent w/v of bovine serum albumin or human albumin. Adjust to pH 8.4 if necessary, using hydrochloric acid.
METHOD
Test solution Dilute the preparation being examined with dilution buffer to obtain a solution containing 0.015 IU of factor II per ml. Prepare at least three further dilutions in dilution buffer.
Reference solution Dilute the reference preparation to be examined with dilution buffer to obtain a solution containing 0.015 IU of factor II per ml. Prepare at least three further dilutions in dilution buffer.
Warm all solutions to 37º in a water-bath shortly before the test.
The following working conditions apply to microtitre plates. If the assay is carried out in tubes, the volumes are adjusted while maintaining the proportions in the mixture.
Using a microtitre plate maintained at 37º, add 25 μl of each dilution of the test solution or the reference solution to each of a series of wells. To each well add 125 μl of dilution buffer, then 25 μl of ecarin and incubate for exactly 2 minutes. To each well add 25 μl of factor IIa chromogenic substrate.
Read the rate of change of absorbance at 405 nm (Appendix 2.2) continuously over a period of 3 minutes and obtain the mean rate of change of absorbance (ΔA/ minute). If continuous monitoring is not possible, read the absorbance at 405 nm at suitable consecutive intervals, for instance 40 seconds, plot the absorbances against time on a linear graph and calculate ΔA/minute as the slope of the line. From the ΔA/minute values of each individual dilution of standard and test preparations, calculate the potency of the preparation being examined and check the validity of the assay by the “Statistical Analysis of Results of Biological Assay and Tests” (Appendix 9).
15.1.8 Biological Assay of Human Coagulation Factor VII
Human coagulation factor VII is assayed by its biological activity as a factor VIIa-tissue factor complex in the activation of factor X in the presence of calcium ions and phospholipids. The potency of a factor VII preparation is estimated by comparing the quantity necessary to achieve a certain rate of factor Xa formation in a test mixture containing the substances that take part in the activation of factor X, and the quantity of the International Standard, or of a reference preparation calibrated in International Units, required to produce the same rate of factor Xa formation.
The chromogenic assay method consists of two consecutive steps: the factor VII-dependent activation of factor X reagent mixture containing tissue factor, phospholipids and calcium ion, followed by enzymatic cleavage of a chromogenic factor Xa substrate into a chromophore that can be quantified spectrophotometrically. Under appropriate assay conditions, there is a linear relation between the rate of factor Xa formation and the factor VII concentration. The assay is summarized in Fig. 1.
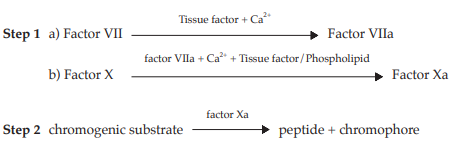
Fig. 1 Schematic Representation of the Assay of Human Coagulation Factor VII
Both steps employ reagents that may be obtained commercially from a variety of sources. Although the composition of individual reagents may be subject to some variation, their essential features are described in the following specification.
REAGENTS
The coagulation factor reagent comprises purified proteins derived from human or bovine sources. These include factor X and thromboplastin tissue factor/ phospholipids as factor VII activator. These proteins are partly purified and do not contain impurities that interfere with the activation of factor VII or factor X. Factor X is present in amounts giving a final concentration during the first step of the assay of 10 to 350 nmol per litre, preferably 14 to 70 nmol per litre. Thromboplastin from natural sources (bovine or rabbit brain) or synthetic preparations may be used as the tissue factor/ phospholipids component. Thromboplastin suitable for use in prothrombin time determination is diluted 1:5 to 1:50 in buffer such that the final concentration of Ca2+ is 15 to 25 nmol per litre. The final factor Xa generation is performed in a solution containing human or bovine albumin at a concentration such that adsorption losses do not occur and which is appropriately buffered at pH 7.3 to 8.0. In the final incubation mixture, factor VII must be the only rate-limiting component and each reagent component must lack the ability to generate factor Xa on its own.
The second step comprises the quantification of the formed factor Xa employing a chromogenic substrate that is specific for factor Xa. Generally this consists of a short peptide of between three and five amino acids, bound to a chromophore group. On cleavage of this group from the peptide substrate, its absorption maximum shifts to a wavelength allowing its spectrophotometric quantification. The substrate is usually dissolved in water and used at a final concentration of 0.2 to 2 nmol per litre. The substrate may also contain appropriate inhibitors to stop further factor Xa generation (addition of edetate).
ASSAY
Reconstitute the entire contents of one ampoule of the reference preparation and the preparation to be examined by adding the appropriate quantity of water; use within 1 hour. Add sufficient prediluent to the reconstituted preparations to produce solutions containing 0.5 to 2.0 IU of factor VII per ml.
(Note Prepare all dilutions in plastic tubes and use within 1 hour.)
Prepare further dilutions of reference and test preparations using an isotonic non-chelating buffer containing 1 per cent of bovine serum albumin or human albumin, buffered preferably between pH 7.3 and 8.0. Prepare at least three separate, independent dilutions for each material, preferably in duplicates. Prepare the dilutions such that the final factor VII concentration is below 0.005 IU per ml.
Prepare a control solution that includes all components except factor VII.
Step 1 Mix dilutions of the factor VII reference preparation and the preparation to be examined with an appropriate volume of the prewarmed coagulation factor reagent or a combination of its separate constituents, and incubate the mixture in plastic tubes or microplate wells at 37º. The concentrations of the various components during the factor Xa generation must be as specified above under the description of the reagents.
Allow the activation of factor X to proceed for a suitable time, usually terminating the reaction before the factor Xa concentration has reached its maximal level in order to obtain a satisfactory linear doseresponse relationship. The activation time is also chosen to achieve linear production of factor Xa in time. Appropriate activation times are usually between 2 and 5 minutes, but deviations are permissible if acceptable linearity of the dose-response relationship is thus obtained.
Step 2 Terminate the activation by the addition of a prewarmed reagent containing a chromogenic substrate. Quantify the rate of substrate cleavage, which must be linear with the concentration of factor Xa formed, by measuring the absorbance change at an appropriate wavelength using a spectrophotometer, either monitoring the absorbance continuously, thus allowing the initial rate of substrate cleavage to be calculated, or terminating the hydrolysis reaction after a suitable interval by lowering the pH by addition of a suitable reagent, such as 50 per cent v/v solution of acetic acid or a 1 M citrate buffer solution pH 3. Adjust the hydrolysis time to achieve a linear development of chromophore with time. Appropriate hydrolysis times are usually between 3 and 15 minutes, but deviations are permissible if better linearity of the dose-response relationship is thus obtained.
Check the validity of the assay and calculate the potency of the test preparation as described in the “Statistical Analysis of Results of Biological Assays and Tests” (Appendix 9).
15.1.9 Biological Assay of Human von Willebrand Factor
The biological functions of human von Willebrand factor are numerous. At present, its ristocetin cofactor activity and its collagen binding activity can be utilized for assays. The potency of human von Willebrand factor is determined by comparing, in given conditions, its activity with the same activity of a reference preparation calibrated against the International Standard, in International Units where applicable.
RISTOCETIN COFACTOR ASSAY
The ristocetin cofactor activity of von Willebrand factor is determined by measuring agglutination of a suspension of platelets in the presence of ristocetin A.
The assay can be carried out for quantitative determinations by using automated instruments, or for semiquantitative determinations by visually assessing the end-point of agglutination in a dilution series. Quantitative assays are preferred.
Reagents
SUSPENSION OF PLATELETS Use standardized and, for example, formaldehyde- or paraformaldehyde-fixed preparations of freshly isolated and washed human platelets. The suspension may also be freeze-dried. An appropriate amount of ristocetin A is added if necessary. Some platelet reagents may already contain ristocetin A.
REFERENCE PREPARATION The reference preparation for von Willebrand factor is the WHO International Standard for von Willebrand factor concentrate.
Method
SEMI-QUANTITATIVE ASSAY Prepare suitable dilutions of the preparation to be examined and of the reference preparation, using as diluent a solution containing 1 to 5 per cent w/v of human albumin in saline TS. Add to each dilution an appropriate amount of the suspension of platelets and, if necessary, of ristocetin A. Mix on a glass slide by moving it gently in circles for 1 minute. Allow to stand for a further 1 minute and read the result against a dark background with side lighting. The last dilution which clearly shows visible agglutination indicates the ristocetin cofactor titre of the sample. Use diluent as a negative control.
QUANTITATIVE ASSAY Reconstitute the entire contents of one ampoule of the reference preparation and the preparation to be examined by adding the appropriate quantity of the recommended diluent (for example water); use immediately. Add sufficient prediluent to the reconstituted preparations to produce solutions containing 0.5 to 2.0 IU per ml. The prediluent consists of an isotonic non-chelating buffer, for example, 1 to 5 per cent of bovine serum albumin or human albumin, and tris(hydroxymethyl)methylamine or imidazole, appropriately buffered.
The test is performed in accordance with the manufacturer’s instructions with at least two dilution series with as many dilutions as are needed to obtain a total of at least three different concentrations in the linear of the assay.
Check the validity of the assay and calculate the potency of the test preparation as described in the “Statistical Analysis of Results of Biological Assay and Tests” (Appendix 9).
COLLAGEN-BINDING ASSAY
Collagen-binding is determined by an enzymelinked immunosorbent assay on collagen-coated microtitre plates. The method is based on the specific binding of von Willebrand factor to collagen fibrils and the subsequent binding of polyclonal anti-von Willebrand factor antibody conjugated to an enzyme, which on addition of a chromogenic substrate yields a product that can be quantitated spectrophotometrically. Under appropriate conditions, there is a linear relationship between von Willebrand factor collagen-binding and absorbance.
Reagents
COLLAGEN Use native equine or human fibrils of collagen type I or III. For ease of handling, collagen solutions may be used.
COLLAGEN DILUENT Dissolve 50 g of dextrose in water, adjust to pH 2.7 to 2.9 with 1 M hydrochloric acid and dilute to 1000 ml with water.
PHOSPHATE-BUFFER SALINE (PBS) Dissolve 8.0 g of sodium chloride, 1.05 g of disodium hydrogenphosphate dihydrate, 0.2 g of sodium dihydrogenphosphate and 0.2 g of potassium chloride in water. Adjust to pH 7.2 using 1 M sodium hydroxide or 1 M hydrochloric acid and dilute to 1000 ml with water.
WASHING BUFFER PBS containing 0.1 per cent w/v solution of polysorbate 20.
BLOCKING REAGENT PBS containing 0.1 per cent w/v of polysorbate 20 and 1 per cent w/v of bovine serum albumin.
DILUTION BUFFER PBS containing 0.1 per cent w/v of polysorbate 20 and 5 per cent w/v of bovine serum albumin.
CONJUGATE Rabbit anti-human von Willebrand factor serum horseradish peroxidase conjugate. Use according to the manufacturer’s instructions.
SUBSTRATE SOLUTION Immediately before use, dissolve a tablet of o-phenylenediamine dihydrochloride and a tablet of urea hydrogen peroxide in 20 ml of water or use a suitable volume of hydrogen peroxide TS (e.g., 1 drop of hydrogen peroxide (10 volumes) TS). Protect from light.
MICROTITRE PLATES Flat-bottomed polystyrene plates with surface properties optimized for enzyme immunoassay and high protein-binding capacity.
Method
TEST SOLUTIONS Reconstitute the preparation to be examined as stated on the label. Dilute with dilution buffer to produce a solution containing approximately 1 IU of von Willebrand factor. Prepare two series of at least three further dilutions using dilution buffer.
REFERENCE SOLUTIONS Reconstitute the reference preparation as directed. Dilute with dilution buffer to produce a solution containing approximately 1 IU of von Willebrand factor. Prepare two series of at least three further dilutions using dilution buffer.
Allow the solution of collagen to warm to room temperature. Dilute with collagen diluent to obtain a solution containing 30 to 75 μg per ml of collagen, and mix gently to produce a uniform suspension of collagen fibrils. Pipette 100 μl into each well of the microtitre plate. Cover the plate with plastic film and incubate at 37º overnight. Empty the wells of the collagen-coated plate by inverting and draining on a paper towel. Add 250 μl of washing buffer. Empty the wells of the plate by inverting and draining on paper towel. Repeat this operation three times. Add 250 μl of blocking reagent to each well, cover the plate with plastic film and incubate at 37º for 1 hour. Empty the wells of the plate by inverting and draining on a paper towel. Add 250 μl of washing buffer. Empty the wells of the plate by inverting and draining on a paper towel. Repeat this operation three times.
Add 100 μl each of the test solutions or reference solutions to the wells of the plate. Add 100 μl of dilution buffer to a series of wells to serve as negative control. Cover the plate with plastic film and incubate at 37º for 2 hours. Empty the wells of the plate by inverting and draining on a paper towel. Add 250 μl of washing buffer. Empty the wells of the plate by inverting and draining on a paper towel. Repeat this operation three times.
Prepare a suitable dilution of the conjugate (for example, a dilution factor of 1 to 4000) with PBS containing 0.5 per cent of bovine serum albumin and add 100 μl to each well. Cover the plate with plastic film and incubate at 37º for 2 hours. Empty the wells of the plate by inverting and draining on a paper towel. Add 250 μl of washing buffer. Empty the wells of the plate by inverting and draining on a paper towel. Repeat this operation three times.
Add 100 μl of substrate solution to each of the wells and incubate at room temperature for 20 minutes in the dark. Add 100 μl of 1 M hydrochloric acid to each of the wells.
Measure the absorbance at the maximum at 492 nm (Appendix 2.2). Use absorbance values to estimate the potency of the preparation to be examined using the “Statistical Analysis of Results of Biological Assay and Tests” (Appendix 9).
The assay is invalid if the absorbances measured for the negative controls are greater than 0.05.
15.1.10 Test for Anti-D Antibodies in Human Immunoglobulin for Intravenous Administration
REAGENTS
Phosphate-buffered saline (PBS) Dissolve 8.0 g of sodium chloride, 0.76 g of anhydrous disodium hydrogenphosphate, 0.2 g of potassium chloride, and 0.2 g of potassium dihydrogenphosphate in water and dilute to 1000 ml with the same solvent. If the solution has to be kept for several days, 0.2 g of sodium azide may be added in order to avoid microbial contamination.
Papain solution Use serological-grade papain from a commercial source, the activity of which has been validated.
Red blood cells Use pooled D-positive red blood cells from not less than three donors, preferably of group OR2R2. D-positive red blood cells may also be obtained from OR1R1 or OR1R2 donors. Mixing phenotypes has not been tested and is therefore not recommended.
Use pooled D-negative red blood cells, preferably from three donors of group Orr. When only one donor of group Orr is available, D-negative red blood cells from only one donor may be used.
Wash the cells four times with PBS or until the supernatant is clear. Centrifuge the cells at 1800 × g for 5 minutes to pack. Treat the packed cells with papain solution according to the manufacturer’s instructions.
Store red blood cells for not more than one week in a preservative solution. A preparation of the following composition is appropriate:
| Trisodium citrate | 8 g | |
| Dextrose | 20 g | |
| Citric acid | 0.5 g | |
| Sodium chloride | 4.2 g | |
| Inosine | 0.938 g | |
| Adenosine triphosphate (ATP) | 0.4 g | |
| Chloramphenicol | 0.34 g | |
| Neomycin sulfate | 0.1 g | |
| Water to | 1000 ml |
Microtitre plates Use V-bottomed rigid microtitre plates.
Reference substances Immunoglobulin (anti-D antibodies test) RS and Immunoglobulin (anti-D antibodies test negative control) RS are suitable for use as the reference preparation and negative control, respectively.
METHOD
The test described in this Appendix is performed at room temperature on the reference solutions, the negative control solutions and the test solutions at the same time and under identical conditions.
Reference solutions and negative control solutions Reconstitute the reference preparation and the negative control according to instructions. The immunoglobulin G (lgG) concentration is 5 per cent w/v solution in each of the reconstituted preparations. Make a twofold dilution of each reconstituted preparation with PBS containing 0.2 per cent w/v solution of bovine serum albumin to give solutions containing 2.5 per cent w/v solution of IgG. Prepare seven further serial twofold dilutions of each preparation using PBS containing 0.2 per cent w/v solution of bovine serum albumin to extend the dilution range to 1/256 (0.0195 per cent w/v solution of IgG). Add 20 μl of each dilution to the microtitre plate.
Test solutions Dilute the preparation to be examined with PBS containing 0.2 per cent w/v solution of bovine serum albumin to give a starting IgG concentration of 2.5 per cent w/v solution. For a 5 per cent w/v solution of products, this is a twofold dilution; adjust the dilution factor accordingly for samples that are not 5 per cent w/v solution to give a starting concentration of 2.5 per cent w/v solution for testing. This 2.5 per cent w/v solution is assigned a nominal twofold dilution factor for comparison with the reference preparations, even if this does not reflect the true dilution factor used to achieve 2.5 per cent w/v solution. Prepare seven further serial twofold dilutions of each preparation using PBS containing 0.2 per cent w/v solution of bovine serum albumin to extend the nominal dilution range to 1/256 (0.0195 per cent w/v solution of IgG) for comparison with the reference preparations over the same IgG concentration range. Make two independent sets of dilutions. Add 20 μl of each dilution to the microtitre plate.
Prepare 3 per cent v/v suspensions of papaintreated D-positive (preferably OR2R2, but OR1R1 or OR1R2 may also be used) and D-negative (Orr) red blood cells in PBS containing 0.2 per cent w/v solution of bovine serum albumin. Add 20 μl of D-positive cells to one dilution series of each of the preparation to be examined, the reference preparation and the negative control, and 20 μl of D-negative cells to the other dilution series of each of the preparation to be examined, the reference preparation and the negative control. Mix by shaking the plate on a shaker for 10 seconds.
Centrifuge the plate at 80 × g for 1 minute to pack the cells. Place the plate at an angle of approximately 70º. Read after at least 3 minutes and once the cells have streamed in the wells containing the negative control and the wells where the D-negative cells have been added. A cell button at the bottom of the well indicates a positive result. A stream of cells represents a negative result.
Record the end-point titre as the reciprocal of the highest dilution that gives rise to a positive result.
The negative control must have a titre not more than two, otherwise an investigation of the test reagents and conditions has to be performed.
The titre of the preparation to be examined is not more than the titre of the reference preparation when all preparations are titrated from a 2.5 per cent w/v solution.
15.1.11 Test for Anticomplementary Activity of Immunoglobulin
For the measurement of anticomplementary activity (ACA) of immunoglobulin, a defined amount of test material (10 mg of immunoglobulin) is incubated with a defined amount of guinea-pig complement (20 CH50) and the remaining complement is titrated; the anticomplementary activity is expressed as the percentage consumption of complement relative to the complement control considered as 100 per cent.
The hemolytic unit of complement activity (CH50) is the amount of complement that, in the given reaction conditions, will produce the lysis of 2.5 × 108 out of a total of 5 × 108 optimally sensitized red blood cells.
REAGENTS
Magnesium and calcium stock solution Dissolve 1.103 g of calcium chloride and 5.083 g of magnesium chloride in water and dilute to 25 ml with the same solvent.
Barbital buffer stock solution Dissolve 207.5 g of sodium chloride and 25.48 g of barbital sodium in 4000 ml of water and adjust to pH 7.3 using 1 M hydrochloric acid. Add 12.5 ml of magnesium and calcium stock solution and dilute to 5000 ml with water. Filter through a membrane filter (pore size 0.22 μm). Store at 4º in glass containers.
Gelatin solution Dissolve 12.5 g of gelatin in about 800 ml of water and heat to boiling in a water-bath. Cool to 20º and dilute to 10 litres with water. Filter through a membrane filter (pore size 0.22 μm). Store at 4º. Use clear solutions only.
Citrate solution Dissolve 8.0 g of sodium citrate, 4.2 g of sodium chloride and 20.5 g of dextrose in 750 ml of water. Adjust to pH 6.1 using a 10 per cent w/v solution of citric acid and dilute to 1000 ml with water.
Gelatin barbital buffer solution Add 4 volumes of gelatin solution to 1 volume of barbital buffer stock solution and mix. Adjust to pH 7.3, if necessary, using 1 M sodium hydroxide or 1 M hydrochloric acid. Maintain at 4º. Prepare fresh solutions daily.
Stabilized sheep blood Collect one volume of sheep blood into one volume of citrate solution and mix. Store at 4º for not less than 7 days and not more than 28 days. (Stabilized sheep blood or sheep red blood cells are available from a number of commercial sources.)
Hemolysin Antiserum against sheep red blood cells prepared in rabbits. (Such antisera are available from a number of commercial sources.)
Guinea-pig complement Prepare a pool of serum from the blood of not less than 10 guinea-pigs. Separate the serum from the clotted blood by centrifugation at about 4º. Store the serum in small amounts below –70º.
METHOD
Preparation of a standardized 5 per cent v/v suspension of sheep red blood cell Separate sheep red blood cells by centrifuging an appropriate volume of stabilized sheep blood and wash the cells at least three times with gelatin barbital buffer solution and prepare as a 5 per cent v/v suspension in the same solution. Measure the cell density of the suspension as follows. Add 0.2 ml to 2.8 ml of water and centrifuge the lysed solution for 5 minutes at 1000 × g; the cell density is suitable if the absorbance (Appendix 2.2) of the supernatant liquid at 541 nm is 0.62±0.01. Correct the cell density by adding gelatin barbital buffer solution according to the expression:
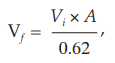
where Vf = final adjusted volume,
Vi = the initial volume, and
A = absorbance of the original suspension at 541 nm.
The adjusted suspension contains about 1 × 109 cells per ml.
HEMOLYSIN TITRATION
Prepare hemolysin dilutions as shown in Table 1.
Table 1
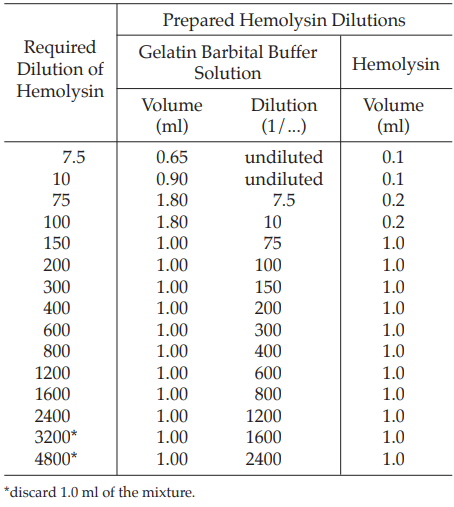
Add 1.0 ml of a 5 per cent v/v suspension of sheep red blood cell to each tube of the hemolysin dilution series, starting at the 1:75 dilution, and mix. Incubate at 37º for 30 minutes.
Transfer 0.2 ml of each of these incubated mixtures to new tubes and add 1.10 ml of gelatin barbital buffer solution and 0.2 ml of diluted guinea-pig complement (for example, 1:150). Perform this in duplicate.
As the unhemolyzed cell control, prepare three tubes with 1.4 ml of gelatin barbital buffer solution and 0.1 ml of a 5 per cent v/v suspension of sheep red blood cell.
As the fully hemolyzed control, prepare three tubes with 1.4 ml of water and 0.1 ml of a 5 per cent v/v suspension of sheep red blood cell.
Incubate all tubes at 37º for 60 minutes and contrifuge at 1000 × g for 5 minutes. Measure the absorbance (Appendix 2.2) of the supernatants at 541 nm and calculate the percentage degree of hemolysis in each tube using the formula:
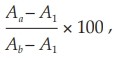
where Aa = absorbance of tubes with hemolysin dilution,
Ab = mean absorbance of the three tubes with full hemolysis, and
A1 = mean absorbance of the three tubes with no hemolysis.
Plot the percentage degree of hemolysis (Y) as the ordinate against the corresponding reciprocal value of the hemolysin dilution as the abscissa on linear graphpaper. Determine the optimal dilution of the hemolysin from the graph by inspection. Select a dilution such that further increase in the amount of hemolysin does not cause appreciable change in the degree of hemolysis. This dilution is defined as one minimal hemolytic unit (1 MHU) in 1.0 ml. The optimal hemolytic hemolysin dilution for preparation of sensitized sheep red blood cells contains 2 MHU per ml.
The hemolysin titration is not valid unless the maximum degree of hemolysis is 50 to 70 per cent. If the maximum degree of hemolysis is not in this range, repeat the titration with more or less diluted complement solution.
PREPARATION OF OPTIMIZED SENSITIZED SHEEP RED BLOOD CELLS (HEMOLYTIC SYSTEM)
Prepare an appropriate volume of diluted hemolysin containing 2 MHU per ml and an equal volume of the standardized 5 per cent v/v suspension of sheep red blood cell. Add the hemolysin dilution to the standardized cell suspension and mix. Incubate at 37º for 15 minutes, store at 2º to 8º and use within 6 hours.
TITRATION OF COMPLEMENT
Prepare an appropriate dilution of complement (for example, 1:250) with gelatin barbital buffer solution and perform the titration in duplicate as shown in Table 2.
Table 2
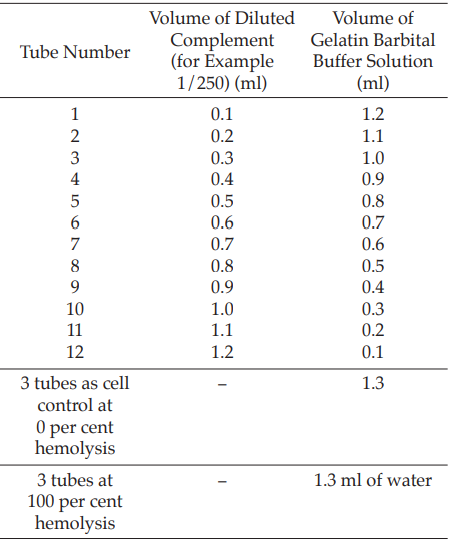
Add 0.2 ml of sensitized sheep red blood cells to each tube, mix well and incubate at 37º for 60 minutes. Cool the tubes in an ice bath and centrifuge at 1000 × g for 5 minutes. Measure the absorbance of the supernatant liquid at 541 nm and calculate the degree of hemolysis (Y) using the formula:
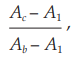
where Ac = absorbance of tubes 1 to 12,
Ab = mean absorbance of tubes with 100 per cent hemolysis, and
A1 = mean absorbance of cell controls with 0 per cent hemolysis.
Plot Y/(1–Y) as the abscissa against the amount of diluted complement in ml as the ordinate on log–log graph paper. Fit the best line to the points and determine the ordinate for the 50 per cent hemolytic complement dose where Y/(1–Y) = 1.0. Calculate the activity in hemolytic units (CH50 per ml) from the formula:
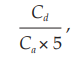
where Cd = reciprocal value of the complement dilution,
Ca = volume of complement in ml resulting in 50 per cent hemolysis, and the scaling factor to take account of the number of red blood cells is 5.
The test is not valid unless the plot is a straight line between 15 per cent and 85 per cent hemolysis and the slope is 0.15 to 0.40, and preferably 0.18 to 0.30.
TEST FOR ANTICOMPLEMENTARY ACTIVITY
Prepare a complement dilution having 100 CH50 per ml by diluting titrated guinea-pig complement with gelatin barbital buffer solution. If necessary, adjust the immunoglobulin being examined to pH 7. Prepare incubation mixtures as shown in Table 3 for an immunoglobulin containing 50 mg per ml.
Table 3
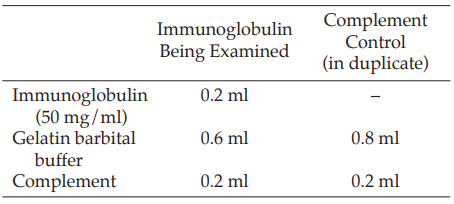
Carry out the test in parallel on the immunoglobulin being examined and prepare ACA negative and positive controls using Human Immunogobulin RS, as indicated in the leaflet accompanying the reference preparation. Higher of lower volumes of sample and of gelatin barbital buffer solution are added if the immunoglobulin concentration varies from 50 mg per ml; for example, 0.47 ml of gelatin barbital buffer solution is added to 0.33 ml of immunoglobulin containing 30 mg per ml to give 0.8 ml. Close the tubes and incubate at 37º for 60 minutes. Add 0.2 ml of each incubation mixture to 9.8 ml of gelatin barbital buffer solution to dilute the complement. Perform complement titrations as described above on each tube to determine the remaining complement activity (see Table 2). Calculate the anticomplementary activity of the preparation being examined relative to the complement control considered as 100 per cent from the formula:
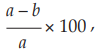
where a = mean complement activity (CH50 per ml) of complement control, and
b = complement activity (CH50 per ml) of tested sample.
The test is not valid unless the anticomplementary activities found for ACA negative control and ACA positive control are within the limits stated in the leaflet accompanying the reference preparation and the complement activity of the complement control (a) is in the range 80 to 120 CH50 per ml.